Nanoparticle Therapy Shows Early Promise at Preventing a Rare, Fatal Newborn Lung Disease
Research By: Fei Sun, PhD | Vlad Kalinichenko, MD, PhD
Post Date: June 11, 2021 | Publish Date: June 11, 2021
If mouse-based results can be translated to human newborns, success could boost nanoparticle therapy for other, more-common lung conditions
The disease is so rare and complex that its acronym is hard to pronounce. But for infants unlucky enough to be born with this lung disease, the outcome is usually fatal.
The disease is called alveolar capillary dysplasia with misalignment of the pulmonary veins (ACDMPV). Research indicates the disease is linked to mutations in the FOXF1 gene. Worldwide, medical experts have documented about 200 cases, but an unknown number of infants may have died without the condition ever being diagnosed, according to the National Organization for Rare Disorders.
The disease is caused by genetic variations that prevent proper blood vessel formation in the lungs. Within days or weeks after birth, infants turn blue from lack of oxygen while blood pressure spikes within their lungs. The few who survive do so by receiving extremely rare infant-sized lung transplants.
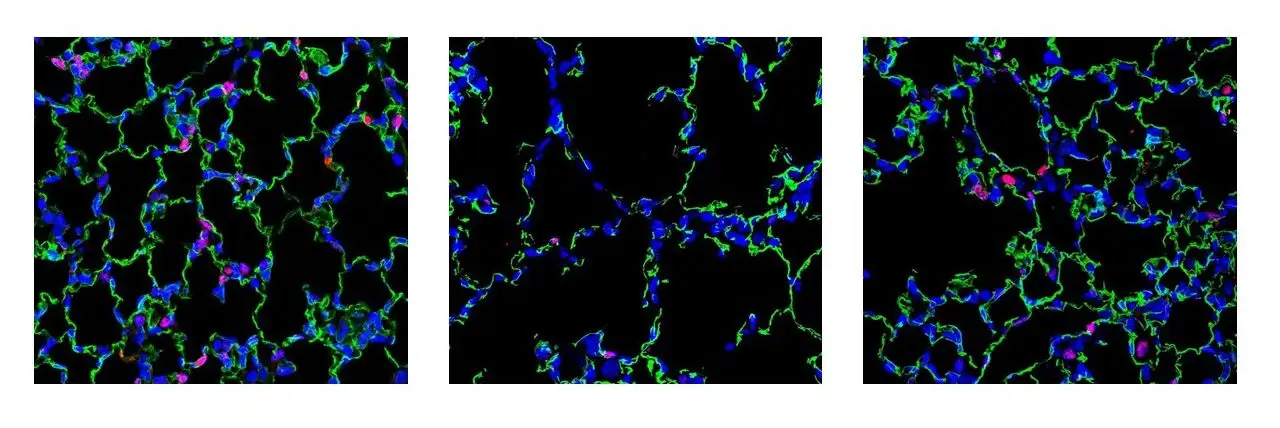
Now, a study led by experts at Cincinnati Children’s and the University of Cincinnati reports helping mice (with a FOXF1 mutation identical to human ACDMPV patients) survive longer with this deadly disease by using high-tech nanoparticles to deliver a STAT3 gene into the lungs to stimulate blood vessel growth. STAT3 is a key downstream target of FOXF1, and its delivery can correct the vascular deficiency in ACDMPV mice. Details were published online June 11, 2021, in the journal Circulation.
If these results can be matched in human studies in the years to come, the co-authors say this success could boost the pace of development for other nanoparticle-based therapies for a wide range of conditions.
“Nanoparticle carriers have shown minimal toxicity and have accelerated the development of novel therapies for human cancers, diabetes and chronic inflammatory disorders. We have developed a unique nanoparticle delivery system that can deliver genes capable of stimulating micro-vessel growth in the newborn lung,” says the study’s senior author Vlad Kalinichenko, MD, PhD, a member of the Center for Lung Regenerative Medicine and the Perinatal Institute at Cincinnati Children’s. “This study shows that a single injection of the nanoparticles with the STAT3 gene vector was sufficient to increase alveolar capillary density, prevent excessively high blood pressures, and dramatically improve survival.”
Without treatment, about 70% of mice born with ACDMPV die within 28 days of birth. The new treatment reduced that mortality rate to 35%, says the study’s first author Fei Sun, PhD, a member of the Center for Lung Regenerative Medicine at Cincinnati Children’s.
Gene-driven therapy but not gene editing
Unlike gene replacement therapies that can make permanent changes to the body, this nanoparticle approach involves materials that do not stay in the body longer than seven days. And yet, in the mice studied, a single treatment early after birth was enough to divert an entire stream of later-developing problems that occur with ACDMPV.
The therapy works by delivering an engineered nanoparticle made of several polymers, fatty acids and a bit of cholesterol that carries the non-integrating STAT3 gene, which in turn prompts blood vessel growth in the lung tissue.
Kalinichenko and colleagues observed the molecular processes involved as part of their ongoing studies of lung development. The nanoparticle was developed with help from Zicheng Deng and Andrew Dunn, who are graduate students mentored by Donglu Shi, PhD, from the Materials Science and Engineering Program at the University of Cincinnati.
With more blood vessels in place, the rapidly growing newborn lungs formed in a closer-to-normal fashion, without setting off dangerous molecular “remodeling” signals that can cause permanent malformations and death from lung failure.
The study details how the treatment improved several measures of lung, heart and blood vessel health, including arterial oxygenation levels, blood pressure in the right ventricle, the ratio of pulmonary acceleration time to pulmonary ejection time (PAT/PET), the diameter of pulmonary arteries, and the thickness of their walls.
Next steps
Much more work must be completed before the nanoparticles can be tried in human newborns with ACDMPV, including safety tests and determining whether repeated treatments would be needed.
The Pediatric Lung Transplant Program at Cincinnati Children’s, which performs transplants on some of the smallest infants, plans to continue working closely with Kalinichenko and colleagues as their studies progress. Some families travel across the globe for care here. Watch this video.
About the study
Co-authors for this study also included Guolun Wang, PhD, Arun Pradhan, PhD, Kui Xu, MD, Jose Gomez-Arroyo, MD, PhD, Yufang Zhang, MS, Gregory Kalin, medical student, Ronald Vagnozzi, PhD, Hua He, PhD, Yuhua Wang, PhD, Allen York, BS, Rashmi Hegde, PhD, Jason Woods, PhD, Tanya Kalin MD, PhD, and Jeffery Molkentin, PhD.
Funding for this work included multiple grants from the National Institutes of Health (HL84151, HL141174, HL149631, HL132849, and HL152094).
| Original title: | Nanoparticle Delivery of STAT3 Alleviates Pulmonary Hypertension in a Mouse Model of Alveolar Capillary Dysplasia |
| Published in: | Circulation |
| Publish date: | June 11, 2021 |
Research By


Latest Posts



About this blog
The Research Horizons blog features news and insights about the latest discoveries and innovations developed by the scientists of Cincinnati Children's. This blog does not provide medical advice, diagnosis, or treatment. Email researchnews@cchmc.org with questions or ideas.
Subscribe to Our Newsletter