Dose Escalation Sharply Improves Hydroxyurea Benefit for Children with Sickle Cell Anemia
Post Date: March 22, 2021 | Publish Date: June 25, 2020

“The study shows clearly that the optimized dosing strategy for hydroxyurea, though it requires more effort than a fixed-dose treatment regimen, results in far better outcomes for children with sickle cell anemia.”
—Russell Ware, MD, PhD
The clinical trial results in Uganda were so clear the study was stopped early.
Rather than using a single, common dosage of the drug hydroxyurea, escalating the dose to a maximum tolerated level significantly lowered the complications from sickle cell anemia.
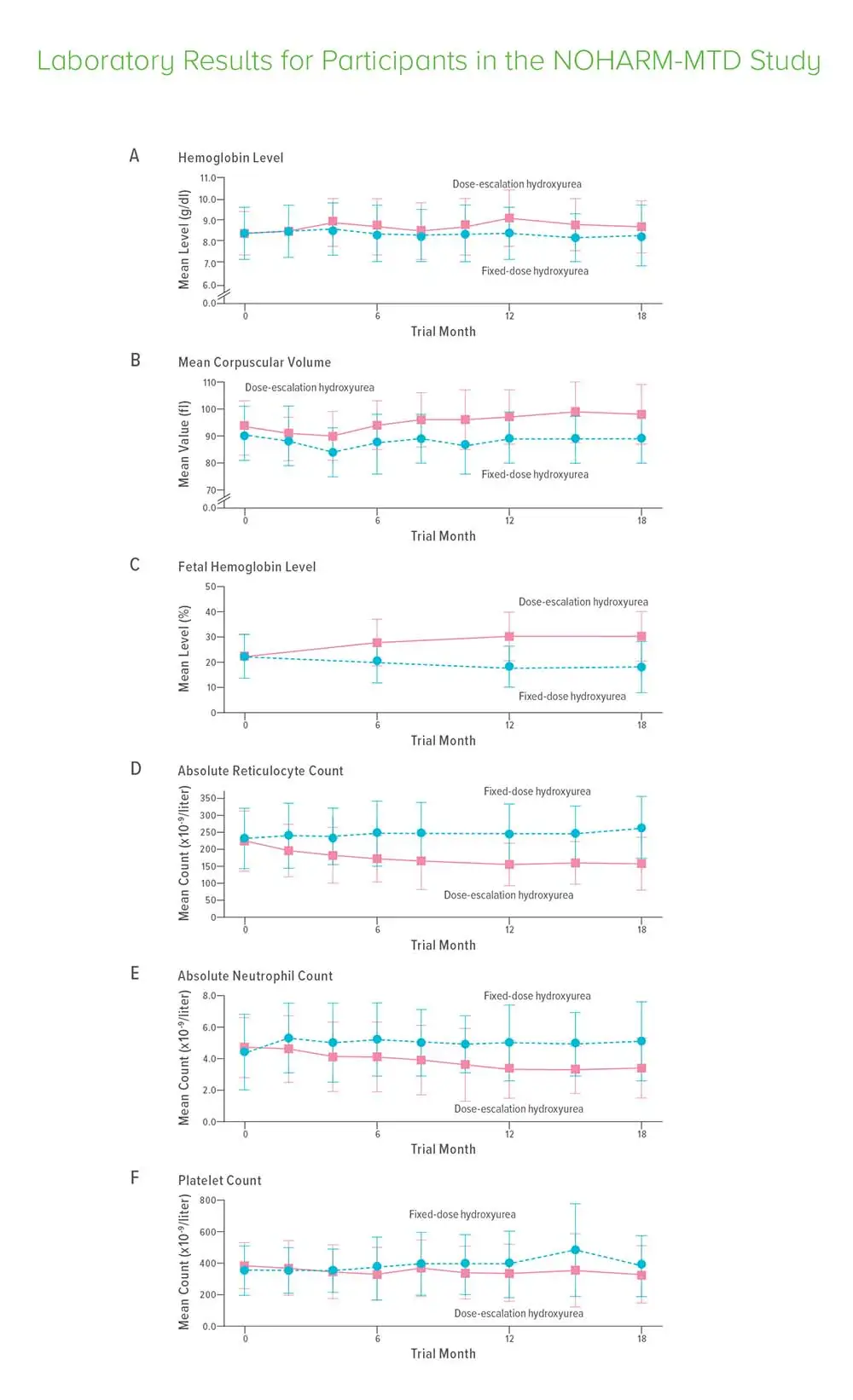
Gradually increasing dosages to about 50% higher than previously used levels reduced hospitalizations by 79%, transfusions by 70%, the risk of acute chest syndrome or pneumonia by 73%, and the risk of a vaso-occlusive pain crisis by 57%, according to data from the NOHARM-MTD study. (Novel use Of Hydroxyurea in an African Region with Malaria – Maximum Tolerated Dose).
The clinical trial results were the latest step forward for a years-long effort led by Russell Ware, MD, PhD, at Cincinnati Children’s and collaborators to demonstrate that hydroxyurea offers an effective, low-cost treatment for sickle cell anemia without increasing malaria risks. Collaborators on the NOHARM-MTD study included experts from Makerere University in Kampala, Uganda, and the Indiana University School of Medicine.
“Our study’s data safety and monitoring board noted a highly significant difference between the treatment groups, with the children on escalated dosing having superior clinical results but the same number of side effects, so at their recommendation we halted the trial and moved all of the children to that escalated dosing strategy,” said Robert Opoka, MMed, who oversaw the study at Makerere University.
HOW THE STUDY WORKED
About half of the 187 study participants received a fixed dose of 20 mg per kilogram of body weight per day. The other half received an escalating dose, which started at 25 mg per kilogram of body weight then increased up to 35 mg per kilogram of body weight per day, if tolerated.
Doctors evaluated the children every 2-3 months. Dose-limiting toxic effects were similar in the two groups, and there were no cases of severe neutropenia or thrombocytopenia.
The study called for tracking the two groups for two years, but the monitoring board opened the dose escalation arm to all participants at about 18 months into the project.
“The study shows clearly that the optimized dosing strategy for hydroxyurea, though it requires more effort than a fixed-dose treatment regimen, results in far better outcomes for children with sickle cell anemia,” Ware says.
LOW-COST TREATMENT FOR CHILDREN IN NEED
Sickle cell anemia affects about 100,000 people in the United States and millions more worldwide. Globally, an estimated 300,000 children are born with sickle cell each year, with about 80% located in sub-Saharan Africa. In the US, most people with sickle cell live well into adulthood, but in low-resource nations the disease kills many children before they reach age 5.
Hydroxyurea works by boosting fetal hemoglobin, Ware says, which reduces sickling in red blood cells, ameliorates anemia, moderates pain and prevents other sickle-related events. The dose escalation approach will come with some higher costs for up-front testing, but the researchers say those costs pale in comparison to the reduction in longer term hospital care. Unlike regular blood transfusions, a long-used treatment in richer countries, hydroxyurea could become accessible to children in any nation.

A LONG JOURNEY FOR A HOPEFUL THERAPY
Cincinnati Children’s co-authors in this NEJM study included Adam Lane, PhD, and Teresa Latham, MA.
For Ware and colleagues, the study caps an effort that includes data from the REACH study, published in the NEJM in December 2018; results from the initial NOHARM study, published in Blood in December 2017; and the US-based BABY HUG trial, with results published in 2011 in The Lancet.
Doctors have used hydroxyurea as an off-label treatment for sickle cell disease since the 1980s. The U.S. Food and Drug Administration (FDA) approved it for treating adults with sickle cell in 1998. It took another 19 years for the FDA to approve hydroxyurea for use in children.
Now, Ware says the next big steps in extending the lifespans of children with sickle cell anemia rest with leaders of health systems in Africa and the global health community who can combine forces to assure that hydroxyurea reaches all the children who need it.

More Top 5 Achievements from the FY20 Research Annual Report:
-
World’s First Three-Organoid System Opens Doors for Medical Research and Diagnosis
-
Cardiac Stem Cell Therapy Improves Scar Formation Rather Than Prompting Cardiomyocyte Regeneration
-
Clinical Trial Success Caps Long Journey for HLH Treatment
-
Single Cell Approach Reveals Impact of Disease-Causing Gene Mutations
Read the full report
Request a PDF
| Original title: | Hydroxyurea Dose Escalation for Sickle Cell Anemia in Sub-Saharan Africa |
| Published in: | The New England Journal of Medicine |
| Publish date: | June 25, 2020 |

Latest Posts
About this blog
The Research Horizons blog features news and insights about the latest discoveries and innovations developed by the scientists of Cincinnati Children's. This blog does not provide medical advice, diagnosis, or treatment. Email researchnews@cchmc.org with questions or ideas.
Subscribe to Our Newsletter