Gene Therapy Restores Key Fragile X Traits in Preclinical Study
Research By: Christina Gross, PhD | Durgesh Tiwari, PhD, M.Pharm | Ernest Pedapati, MD, MS | Craig Erickson, MD
Post Date: July 14, 2026 | Publish Date: July 10, 2026
Cincinnati Children’s-led team reports AAV-based FMR1 strategy improved seizure susceptibility, repetitive behavior and brain activity markers in mice
A gene therapy designed to replace the missing protein that causes fragile X syndrome restored several disease-relevant traits in a mouse model, according to a new study published in Gene Therapy.
Fragile X syndrome is the most common inherited form of intellectual disability and a leading single-gene condition associated with autism. There is no cure, and current care focuses on managing symptoms such as anxiety, sensory sensitivity, hyperactivity, developmental seizures and learning challenges.
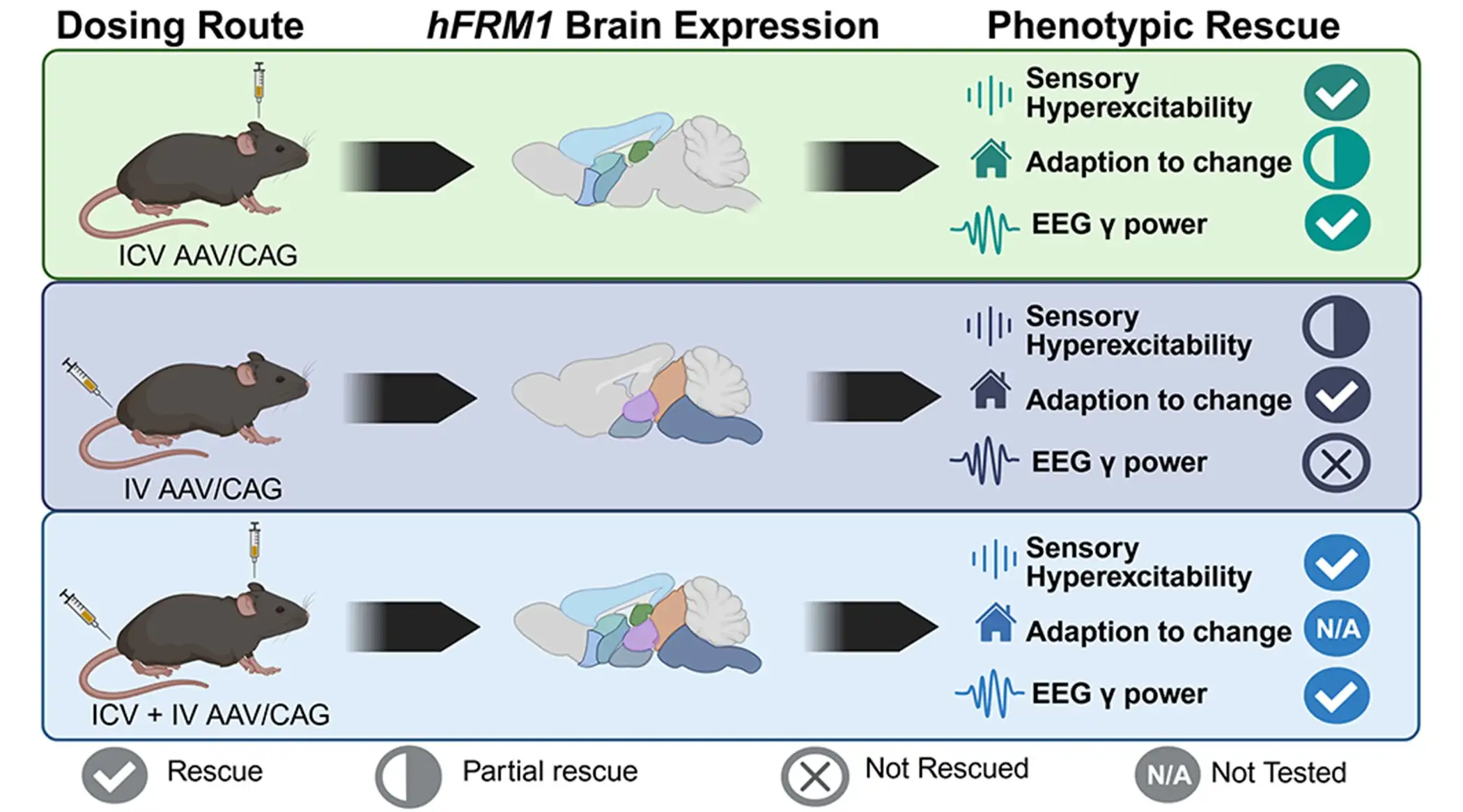
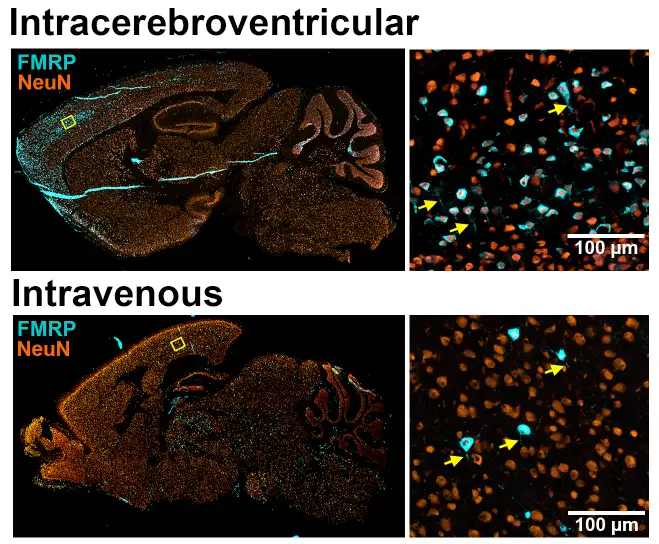
The study, led by investigators at Cincinnati Children’s and collaborators at Forge Biologics, tested adeno-associated viral vectors carrying human FMR1, the gene silenced in fragile X syndrome. After testing several candidates, the team found an approach that produced the FMRP protein in key brain regions and improved multiple phenotypes in Fmr1 knockout mice.
The improvements included reduced susceptibility to audiogenic seizures, improvements in sensory hyperactivity and repetitive digging behavior, and normalization of elevated low-gamma EEG power–a brain activity pattern found in human fragile X studies.
“These findings are important because they show that restoring FMRP can improve several fragile X-related traits in a model designed with clinical translation in mind,” says Christina Gross, PhD, co-corresponding author and researcher in the Division of Neurology at Cincinnati Children’s. “By pairing gene replacement with outcomes that can help bridge mouse studies and future human trials, this work gives the field a stronger foundation for developing therapies that address the root biology of fragile X syndrome.”
Cincinnati Children’s scientists Craig Erickson, MD, MA, Ernest Pedapati, MD, MS, and Durgesh Tiwari, PhD, M.Pharm, also served as co-corresponding authors.
The findings move beyond proof that FMRP can be re-expressed. The study explores delivery routes, promoters, dosing strategies, and other factors that may help define what “translation-ready” preclinical evidence should look like for fragile X gene therapy. The results also reinforce the value of EEG measures as biomarkers that could bridge animal studies and future human trials.
For investors and philanthropists, the work highlights a path toward disease-modifying treatment in an area with high unmet need and no approved therapy. The findings support continued investment in vector design, safety testing, biomarker development and the scalable manufacturing that would be needed before human clinical trials could begin.
For practicing clinicians, this preclinical study does not change current patient care. However, it suggests that a safe method of restoring FMRP may eventually improve clinical concerns that have been difficult to manage otherwise. Importantly, the gene therapy showed benefits even when delivered at different age points. Also, the team details two administration pathways that could be combined to assure the therapy reaches all key parts of the brain.
“Our studies show that re-expression of FMRP in mice at ages equivalent to 4-6 and 15-30 years in humans has the potential to rescue sensory hypersensitivity, stereotypic behavior, and excessive EEG gamma power,” Pedapati says. “This suggests that certain FXS-related deficits are reversible or can be improved by re-expression of FMRP after large parts of brain development have already occurred.”
For people affected by fragile X syndrome and their families, the study offers cautious hope. It suggests that replacing the missing gene product may be feasible and biologically meaningful, but the approach has not yet been tested in people. More research is needed to evaluate safety, durability, dosing, immune responses and the best timing for treatment.
About the study
Richard Lacher, Division of Child and Adolescent Psychiatry, was lead author. Other co-authors from Cincinnati Children’s included Lindsay Wathen, MS, McKenzie Rice, BS, Heather Carles, BS, Angela White, PhD, Austen Fisher, BS, Kaitlin Bucher, BS, Grace Westerkamp, BS, Adam Fritz, BS, Brooke Gollaway, BS, Sebastian Piloto, J. Elliott Robinson, MD, PhD, Michael Williams, PhD, and Charles Vorhees, PhD.
Co-authors with Forge Biologics included Kari Henson, PhD, Caitlin Jones, PhD, Tiffany Arnold, Elizabeth Ramsuchit, Darren Murrey, Rebecca Raig, David Dismuke, PhD, and Erandi K. De Silva.
Funding sources for this work include a research contract through Forge Biologics, a FRAXA fellowship, a National Institutes of Health grant (UL1TR001425), plus an Innovation Funds Award and a CpG award from Cincinnati Children’s.
Erickson, Pedapati, Gross, Dismuke and De Silva note they are shown as co-inventors on Patent Application PCT/US2021/041975.
| Original title: | FMR1 gene therapy restores translationally relevant phenotypes in a mouse model for fragile X syndrome |
| Published in: | Gene Therapy |
| Publish date: | July 10, 2026 |
Research By

The overall goal of the Gross Lab is to identify and study shared pathological mechanisms underlying brain disorders like epilepsy and autism of different etiologies, and to use this knowledge to develop therapeutic treatment approaches.




Latest Posts
About this blog
The Research Horizons blog features news and insights about the latest discoveries and innovations developed by the scientists of Cincinnati Children's. This blog does not provide medical advice, diagnosis, or treatment. Email researchnews@cchmc.org with questions or ideas.
Subscribe to Our Newsletter