From 4,500 Possibilities, One Compound Emerges as Promising Treatment for PAH
Research By: Mingxia Gu, MD, PhD
Post Date: May 4, 2021 | Publish Date: May 5, 2021
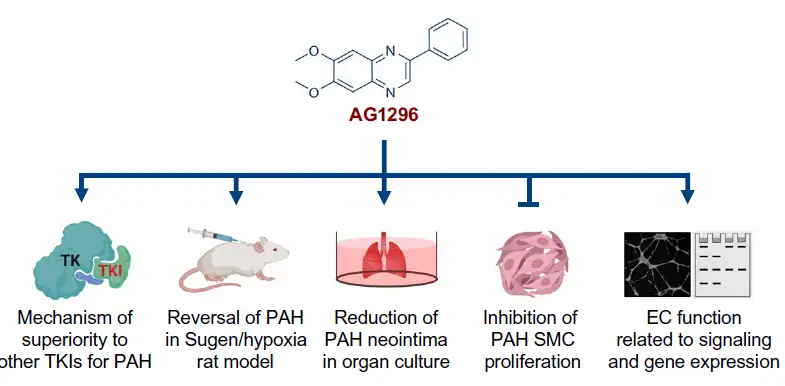
Team from Stanford and Cincinnati Children’s uses patient-derived stem cells as screening tool to identify AG1296 as superior candidate for controlling dangerous pulmonary arterial hypertension
One of the dangerous health conditions that can occur among premature newborns, children born with heart defects, and some others is pulmonary arterial hypertension (PAH).
Commonly mistaken for asthma, this condition occurs when blood vessels in the lungs develop excessive resistance to blood flow. This forces the heart’s right ventricle to work harder, causing it to enlarge, thicken and further elevate blood pressure. While early treatment usually succeeds, the condition can become persistent and progressive, which can lead to heart failure and death.
The exact incidence and prevalence of PAH remains unclear, but reviews of patient registries in Europe have estimated that the condition occurs in nearly 64 of every 1 million children, including transient cases. So far, there is no cure.
Now a team of scientists at Stanford University and Cincinnati Children’s report finding a potentially effective needle in a haystack.
“In this study, we screened a library of 4500 compounds at different stages of clinical development in eight different doses. Of the six compounds that improved cell survival, we identified the tyrosine kinase inhibitor AG1296 as the lead compound for further investigation,” says Mingxia Gu, MD, PhD, lead author of a study published May 5, 2021, in Science Translational Medicine.
This compound has been available for research for several years but has not been approved for medical use. Its potential value as a PAH treatment emerged only after testing an entire library of compounds using several lines of induced pluripotent stem cells (iPSCs) grown to reflect the genetic signatures of actual patients with the condition.
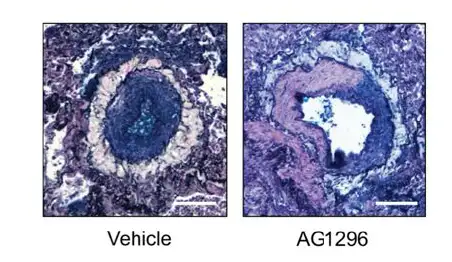
The researchers found that AG1296 outperformed several other TKI inhibitors on a series of gene expression, cell signaling and mechanistic measures. Most importantly, the compound reversed key biomarkers of the disease in human cells and improved vascular function in animal models.
Center of Excellence
The Pulmonary Hypertension Clinic at Cincinnati Children’s is one of eight pediatric centers in the nation to earn accreditation from the Pulmonary Hypertension Association (PHA) as a Pulmonary Hypertension Accredited Comprehensive Care Center.
Cincinnati Children’s also hosts the PAH Biobank, an NIH/NHLBI funded resource of biological samples, genetic data, and clinical data for the PAH research community.
Next Steps
The use of human, PAH-specific stem cells as a drug screening platform reflects a crucial step forward, Gu says. In previous studies, about 90% of potential treatments that showed promise in animal studies failed to show benefits in human clinical trials.
“This is the first time for using iPSCs to screen for compounds for PAH, but people have been using similar strategies to screen for new therapies for other conditions,” Gu says. “Overall, using patient-specific iPSCs for developing precision medicine is a new and promising field.”
Launching a clinical trial for AG1296 likely will require a year or more of preparation and safety evaluations, Gu says. If a trial is launched, it could take a few more years to complete. Then an application process to seek approval from the U.S. Food and Drug Administration would begin.
Learn more about pulmonary hypertension
| Original title: | iPSC–endothelial cell phenotypic drug screening and in silico analyses identify tyrphostin-AG1296 for pulmonary arterial hypertension |
| Published in: | Science Translational Medicine |
| Publish date: | May 5, 2021 |
Research By

Latest Posts

About this blog
The Research Horizons blog features news and insights about the latest discoveries and innovations developed by the scientists of Cincinnati Children's. This blog does not provide medical advice, diagnosis, or treatment. Email researchnews@cchmc.org with questions or ideas.
Subscribe to Our Newsletter