Targeting C5ar1 Shows Early Promise as New Therapy for Gaucher Disease
Research By: Manoj Pandey, PhD
Post Date: June 29, 2019 | Publish Date: March 2, 2017
“We suggest that targeting a molecule called C5aR1 may serve as a viable treatment option for patients with Gaucher disease and possibly other LSDs.”
Blocking a molecule that drives inflammation and organ damage in Gaucher disease and other lysosomal storage diseases (lsds) shows potential as less risky, lower-cost alternative to current therapies, according to findings recently published in Nature.
An international research team from Cincinnati Children’s and the University of Lübeck in Germany conducted a series of experiments using mouse models and human blood samples that took a fresh look at large group of rare diseases. Individually, the 50 genetic diseases characterized as LSDs are rare. Collectively, they occur once in every 8,000 births, making LSDs a major challenge for the healthcare system, according to the National Institutes of Health.
“Current enzyme replacement and substrate reduction therapies are expensive and still associated with inflammation, increased risk of malignancies and Parkinson’s disease,” says study first author Manoj Pandey, PhD, Division of Human Genetics.
People with LSDs lack enzymes that break down used-up proteins and other spent particles, preventing their cells from shedding these waste materials. Current treatments attempt to protect normal cell function by breaking down certain fatty molecules and preventing accumulation of other waste particles.
“We suggest that targeting a molecule called C5aR1 may serve as a viable treatment option for patients with Gaucher disease and possibly other LSDs,” Pandey says.
Study Identifies Critical Part of Inflammatory Process
C5aR1 is a critical part of a molecular pathway that drives pro-inflammatory processes initiated by mutations in the GBA1 gene. GBA1 encodes the lysosomal enzyme glucocerebrosidase (GCase), which degrades the fatty molecule glucosylceramide (GC). C5aR1 is a receptor for a small peptide derived from C5a, a part of the complement system that drives inflammation in several different types of immune cells.
The Gaucher disease process starts by GBA1 mutation driving extensive GC accumulation in immune cells. Before this study, the molecular process that connects GC accumulation to inflammation was unknown.
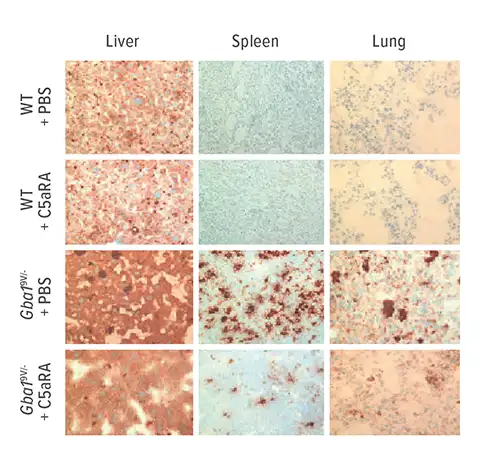
Pandey and colleagues show that inflammatory GC accumulation in spleen, liver, lung and bone marrow immune cells drives the induction of auto-antibodies against GC in mouse models of the disease, which form immune complexes. These immune complexes promote the production of C5a and activation of its receptor. C5aR1 activation essentially tips the balance between GC formation and degradation.
The researchers also found similar evidence of C5aR1 activity in human cells donated by Gaucher patients.
“We propose a dual role model for the C5a–C5aR1 axis in Gaucher disease. First, it serves as a critical pathway promoting the excessive cellular accumulation and release of GC as the basis for the formation of GC-specific immune complexes,” the co-authors wrote. “Such immune complexes then drive extensive complement activation leading to excessive systemic and tissue C5a generation. Second, this excess C5a activates antigen-presenting cells and drives TH1/TH17-cell differentiation resulting in undesired production of pro-inflammatory cytokines and chemokines, eventually causing increased tissue recruitment of activated immune cells that lead to and/or propagate the molecular mechanisms of tissue damage and the resultant Gaucher disease manifestations.”
C5aRA Antagonist Developed
The research team then injected mice with a C5aR antagonist (patent owned by Cincinnati Children’s). They found substantial reduction in macrophage infiltration in lungs, livers and spleens of mice and near-elimination of GC accumulation.
The research team says more study is needed before any clinical trials can begin. That work includes continued testing to determine how the C5aRA molecule works against other LSDs. The team also plans to evaluate the effectiveness of eculizumab, an anti-C5 monoclonal antibody produced by Alexion Pharmaceuticals (which helped fund the current study), as a potential treatment option.
View the entire 2017 Research Annual Report
| Original title: | Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease |
| Published in: | Nature |
| Publish date: | March 2, 2017 |
Research By


Latest Posts


About this blog
The Research Horizons blog features news and insights about the latest discoveries and innovations developed by the scientists of Cincinnati Children's. This blog does not provide medical advice, diagnosis, or treatment. Email researchnews@cchmc.org with questions or ideas.
Subscribe to Our Newsletter