Single-Cell Analysis Sheds New Light on the Rare Lung Disease LAM
Research By: Yan Xu, PhD
Post Date: July 2, 2020 | Publish Date: June 30, 2020
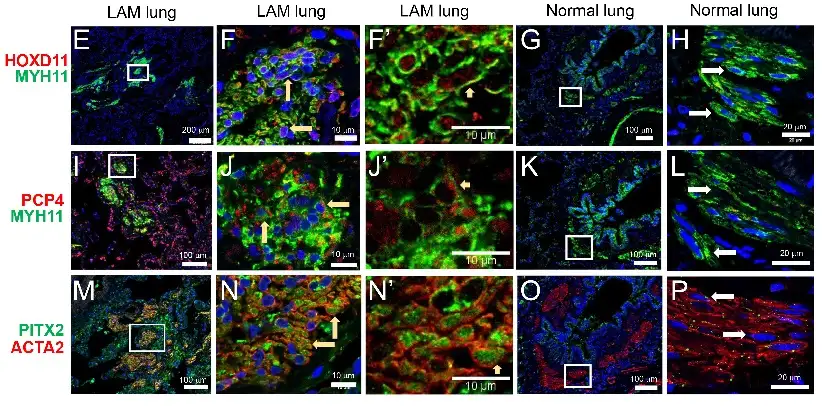
Until recent years, lymphangioleiomyomatosis (LAM) was largely a mystery. This rare lung disease occurs almost exclusively in women and usually strikes during the prime of their lives.
What causes this sudden onset?
Using a single-cell transcriptomic approach, researchers at Cincinnati Children’s and the University of Cincinnati have unlocked new clues to help us understand the origins of LAM. Their findings were published June 30, 2020, in the American Journal of Respiratory and Critical Care Medicine.
For many patients, LAM begins long before they discover its name. The disease causes abnormal smooth muscle cells to grow in the lungs, lymphatic system, and kidneys. Unregulated growth of these cells can lead to severe effects—loss of lung function, accumulation of lymph-rich fluid in the chest and abdomen, and tumor growth in the kidneys.
Because LAM is difficult to detect, and most patients are young—the average age at diagnosis is 35—symptoms often are mistaken for asthma, emphysema, or bronchitis. When patients finally receive an accurate diagnosis, many questions linger. Why did they develop LAM? How will it progress? What is the best treatment?
Learning more about LAM cells can bring us closer to the answers. But these cells are challenging to study—they are difficult to isolate, grow poorly, and lose unique features in culture. This has limited our ability to understand how they function and interact. With advances in single-cell RNA-sequencing technology, the research team identified a new opportunity.
“Insight on LAM cells has been restricted to histological and immunohistochemical studies,” says Yan Xu, PhD, corresponding author and faculty member of the Divisions of Pulmonary Biology and Biomedical Informatics at Cincinnati Children’s. “To our knowledge, this is the first comprehensive study of the LAM lung and uterus using the single-cell transcriptomic approach.”
By creating detailed maps of individual LAM cells, researchers were able to explore each cell at a deeper level than ever before. This led them to discover something completely unique to LAM patients—a new cell type called LAMCORE. These cells are distinct from, but closely related to, normal lung mesenchymal cells, which are important regulators of tissue repair and regeneration.
With uncertainty surrounding the origins of LAM cells, researchers faced obstacles to developing new therapies. In this study, the team discovered that LAM cells in the lung and uterus are morphologically indistinguishable, sharing similar gene expression profiles and biallelic TSC2 mutations. This evidence supports a potential uterine origin for the LAMCORE cell.
What are the genes and processes that are active in these LAMCORE cells, and how do they influence pulmonary cells? To find out, researchers used single-cell RNA sequencing to identify secretome proteins that are present in LAM patient lungs, but not in normal control lungs. These proteins tell us more about how LAM cells cause tissue destruction and affect the cells around them. As a result, they could serve as new biomarkers for diagnosis, prognosis, and therapeutic responses.
“These findings could also have implications beyond LAM,” says Francis McCormack, MD, co-corresponding author and director of the Division of Pulmonary, Critical Care, and Sleep Medicine at the University of Cincinnati. “As we understand more about LAM pathogenesis as a unique model of malignancy, this may yield broader insights into the biology of cancer.”
Learn more about the Xu Lab’s research in single-cell applications in LAM.
| Original title: | Single Cell Transcriptomic Analysis Identifies a Unique Pulmonary Lymphangioleiomyomatosis Cell |
| Published in: | American Journal of Respiratory and Critical Care Medicine |
| Publish date: | June 30, 2020 |
Research By


Latest Posts
About this blog
The Research Horizons blog features news and insights about the latest discoveries and innovations developed by the scientists of Cincinnati Children's. This blog does not provide medical advice, diagnosis, or treatment. Email researchnews@cchmc.org with questions or ideas.
Subscribe to Our Newsletter