Revealing the Molecular Secrets of Trachea-Esophageal Birth Defects
Research By: Aaron Zorn, PhD
Post Date: December 10, 2019 | Publish Date: Dec. 5, 2019
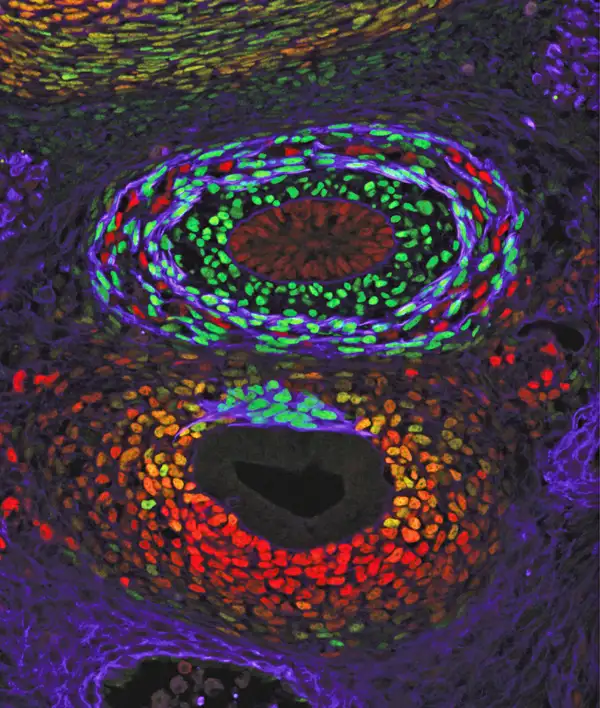
When a baby is born with a tracheoesophageal birth defect, the abnormally connected esophagus and trachea interfere with feeding and the child’s breathing. The birth defects can be fatal and the causes remain a mystery.
A scientist in a Cincinnati Children’s-led research collaborative, funded by the NIH to discover the genetic causes and new treatments for the birth defects, recently published new foundational research in Developmental Cell that provides some new answers.
Developmental biologist Aaron Zorn, PhD, is part of the CLEAR Consortium (Congenital Esophageal and Airway Defect Research). Zorn and his research team used mouse and frog laboratory animals to uncover the biomechanics and molecular pathways of how the trachea and esophagus form and how mutations can cause birth defects.
In fact, they identified four major steps to the process—starting with basic patterning of the fetal gut tube to a final step involving formation of separate trachea and esophagus tubes.
The investigators also found out what can happen when those normal processes go wrong, and they learned new information that might one day help form the basis for developing regenerative therapies, according to Zorn, director of the Center for Stem Cell and Organoid Medicine at Cincinnati Children’s.
The basic genetic and biological processes the researchers found in their mouse and frog models are considered evolutionarily conserved, according to study authors.
“This means the genes we identified as important to tracheoesophageal development in animals remain essentially unchanged throughout the evolution of different species, from the frog to humans,” Zorn explained. “These are genes that evolution can’t tinker with because the changes could be lethal.”
From the Beginning
During the early development of a fetus, the trachea and esophagus arise from the separation of a common foregut tube. The foregut is the anterior part of the gut near the mouth.
Helping create this separation are cellular processes linked to the genes sonic hedgehog (SHH) and glioma-associated oncogene (GLI). The pathway transmits information to embryonic cells that is required for the cells to transform and differentiate into specific cell types, like a trachea or an esophagus.
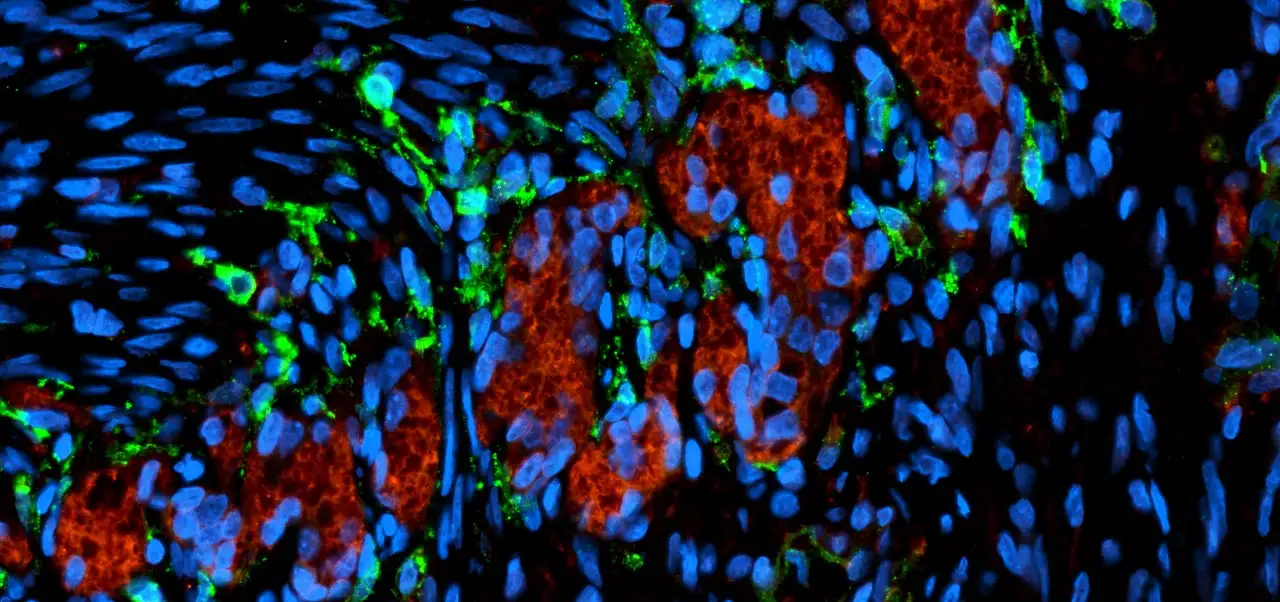
Zorn’s team observed up close how the SHH/GLI pathway coordinates the formation of a trachea and an esophagus in the frog and mouse models. At the same time, they identified additional molecular processes downstream from SHH/GLI, such as Sox2 in the esophagus and Nkx2-1+ in the trachea. Sox2 and Nkx2-1+ also work in unison to form a boundary between the esophagus and trachea that also fuses to form a transient septum.
The researchers also discovered that the formation of the septum and distinct trachea and esophagus tubes requires additional cellular processes and changes that involve GTPase (which acts as a molecular switch) and Rab11. Rab11 is downstream molecule in the GTPase pathway that cycles on and off to help control certain molecular processes.
Nature Interrupted
By using pharmacologic disruption and gene mutations of the SHH/GLI pathway and certain downstream patterning genes, the researchers report these caused tracheoesophageal birth defects in the mouse models.
“This study provides a cellular explanation for earlier mouse models of tracheoesophageal separations and the results also help explain the development of tracheoesophageal birth defects in patients who have SHH and GLI mutations,” Zorn said.
The Cincinnati Children’s investigators who worked on the paper are leading groundbreaking research in using pluripotent stem cells to grow human tissues and miniature human organs called organoids. Although human organoids were not part of the current study, the data it generated in animal models often helps form the scientific basis of future organoid studies.
The study’s first author was Talia Nasr, a graduate student in the Zorn laboratory. A good portion of the study’s supporting data is vividly illustrated in detailed confocal microscopic images produced by the Confocal Imaging Core at Cincinnati Children’s, led by study co-author Matthew Kofron, PhD.
Funding support came in part from the National Institutes of Health (P01HD093363, P30 DL0778392, F30HL142201, T32GM063483-14).
—Article written by Nick Miller
| Original title: | Endosome-Mediated Epithelial Remodeling Downstream of Hedgehog-Gli Is Required for Tracheoesophageal Separation |
| Published in: | Developmental Cell |
| Publish date: | Dec. 5, 2019 |
Research By


Latest Posts

About this blog
The Research Horizons blog features news and insights about the latest discoveries and innovations developed by the scientists of Cincinnati Children's. This blog does not provide medical advice, diagnosis, or treatment. Email researchnews@cchmc.org with questions or ideas.
Subscribe to Our Newsletter