Pulmonary Fibrosis Treatment Shows Proof of Principle
Research By: Satish Madala, PhD | Anil Goud Jegga, DVM, MRes,
Post Date: August 6, 2020 | Publish Date: Aug. 7, 2020
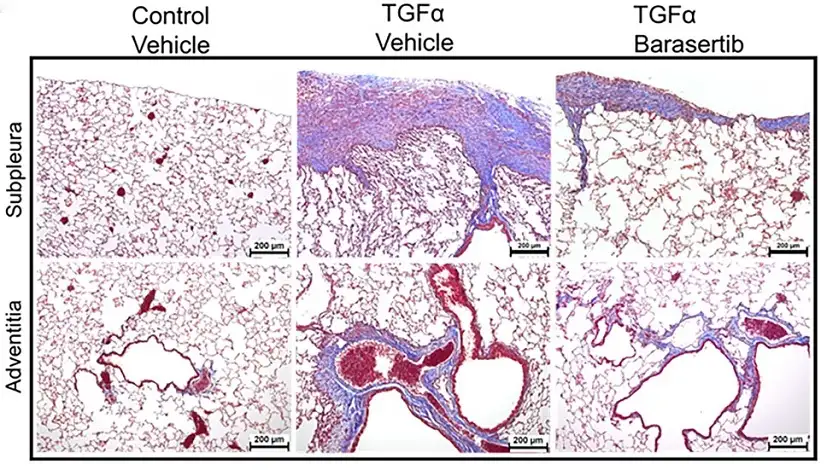
In mice, barasertib stops dangerous scar formation in the lungs
A pre-clinical study led by scientists at Cincinnati Children’s demonstrates that, in mice, the drug barasertib reverses the activation of fibroblasts that cause dangerous scar tissue to build up in the lungs of people with idiopathic pulmonary fibrosis (IPF).
Findings were posted online Aug. 6, 2020, in the journal EMBO Molecular Medicine. The study was led by Rajesh Kasam, PhD, Division of Pulmonary Medicine, and co-corresponding authors Satish Madala, PhD, Division of Pulmonary Medicine, and Anil Goud Jegga, DVM, MRes, Division of Biomedical Informatics.
The discovery suggests that a powerful treatment for a fatal disease that currently has no cure other than lung transplantation may be within a few years of launching human clinical trials.
“This study is the first to identify barasertib as an anti-fibrotic candidate,” Madala says. “That’s important because so far there are no treatments for IPF that appear to reverse the underlying process that causes the disease.”
What is IPF?
Idiopathic pulmonary fibrosis is a chronic, progressive lung disease that usually affects people between the ages of 50 and 70, according to the National Institutes of Health. About 100,000 people in the US have IPF with 30,000 to 40,000 new cases diagnosed each year. Survival time varies, but most people live about 3 to 5 years after diagnosis.
Scientists believe a combination of genetic predisposition and environmental factors trigger IPF, but the exact causes and mechanisms remain unknown.
Currently, there are two approved drugs that can help slow the disease; Ofev (nintedanib) and Esbriet (pirfenidone). But both can cause have serious side effects. Doctors also use oxygen supplementation and other symptom management methods to prolong survival. Ultimately, however, people with IPF have needed lung transplantation, but the supply of donor organs is limited.
Scientists at Cincinnati Children’s have been studying this disease because lung scarring also occurs in children.
“Pulmonary fibrosis is a major cause of death in both adult and pediatric chronic lung disease,” Madala says. “Also, several genetic mutations have been identified in pediatric populations that have been shown to cause familial IPF. Identifying anti-fibrotic therapies will help to treat multiple chronic fibrotic lung diseases.”
New clues emerge about how fibrosis occurs
In the new study, the co-authors reveal that a gene called aurora kinase B (AURKB) is expressed in high levels within the cells of lung scar tissue (also known as fibroblasts). This gene expression appears to be driven by multiple growth factors and a transcription factor called Wilms Tumor 1, the study states.
The team used several technologies to hunt through hundreds of possibilities to find this genetic connection, including gene expression data from IPF patients, transcriptional signatures from approved and investigational drug-treated cells, and a software tool developed at Cincinnati Children’s called ToppFun. Once the team identified the involvement of barasertib, a known AURKB inhibitor, they explored the therapeutic relevance, underlying mechanistics and target pathways related to pulmonary fibrosis.
What is Barasertib?
Barasertib, made by AstraZeneca Pharmaceuticals, is a powerful aurora kinase inhibitor that has shown promise as a potential cancer treatment. In fact, the drug is being studied in Phase I/II clinical trials as a possible treatment for acute myeloid leukemia (AML).
The collaborators in this study used the drug to treat mice that were induced to develop lung disease that mimics human IPF. They found that inhibiting AURKB activity helped the mice in multiple ways.
Those treated with barasertib before developing lung fibrosis were less likely to do so once scar formation was induced. Once fibrosis had started, introducing the drug significantly slowed further disease progress, primarily by causing faster cell death among the fibroblasts. The results of treatment included less scar tissue, improved lung elasticity, and overall better lung function.
“These findings have therapeutic implications as they suggest targeting AURKB activity inhibits fibroblast activation, particularly when multiple pro-fibrotic growth factors promoting alternative fibroproliferative pathways are present,” the co-authors state. “Together, our preclinical studies provide important proof-of-concept that demonstrate barasertib as a possible intervention therapy for IPF.”
What’s next?
“Since the current project was conducted in mouse models and human cells derived from lung biopsies of IPF patients, additional studies are needed to determine whether targeting AURKB would be effective and safe enough to test in human patients,” Jegga says.
Also, barasertib is known to have systemic exposure-related side effects. Therefore, as part of ongoing and future work, the researchers are exploring alternative routes of administration to bypass systemic exposure and ensure targeted delivery.
About the study
This study was supported in part by the NIH (HL134801, R21 AG059533, UG3TR002612) and the US Department of Defense (W81XWH-17-1-0666)
| Original title: | Inhibition of Aurora Kinase B attenuates fibroblast activation and pulmonary fibrosis |
| Published in: | EMBO Molecular Medicine |
| Publish date: | Aug. 7, 2020 |
Research By


Latest Posts

About this blog
The Research Horizons blog features news and insights about the latest discoveries and innovations developed by the scientists of Cincinnati Children's. This blog does not provide medical advice, diagnosis, or treatment. Email researchnews@cchmc.org with questions or ideas.
Subscribe to Our Newsletter