Mitochondria Pore Emerges as Potential Key to Managing Muscular Dystrophies
Research By: Jeffery Molkentin, PhD
Post Date: August 25, 2023 | Publish Date: Aug. 25, 2023
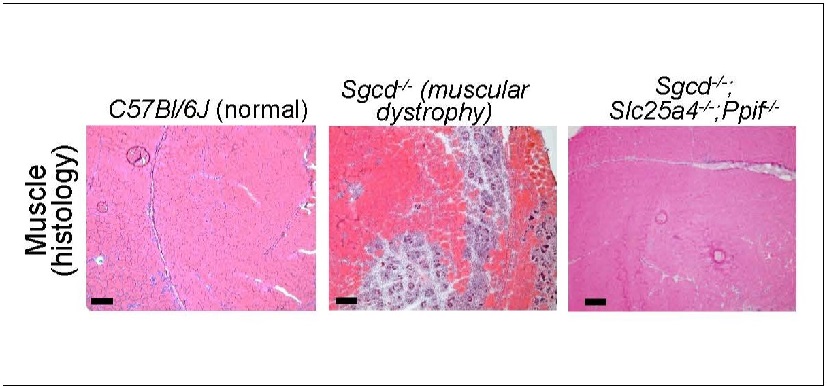
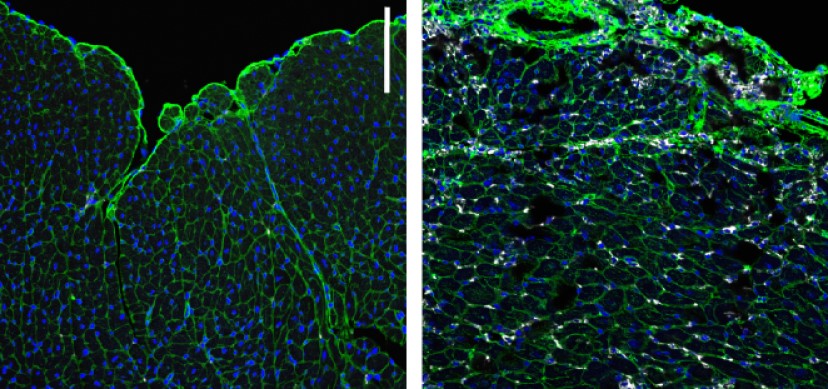
Cincinnati Children’s scientists discover how damage to the cell’s energy factory triggers muscle wasting outcomes. In gene-edited mice, closing a pore in the mitochondrial membrane stops disease progression
Ever since the Jerry Lewis telethons began in the 1960s, millions of people have become familiar with an otherwise rare disease called muscular dystrophy (MD).
The medical world has learned much over the ensuing years, including that more than 30 closely related disorders exist that can produce the gradual muscle degeneration that steals a child’s ability to walk and eventually disrupts other organ functions. An estimated 250,000 people in the U.S. are living with a muscular dystrophy. While many are living longer lives thanks to improved treatments, no cure has been found.
Now an eye-opening study led by scientists at Cincinnati Children’s–published Aug. 25, 2023, in Science Advances–reports an entirely new approach to preventing the muscle-wasting symptoms of MD. The research focuses on the role played by mitochondria, the tiny organelle within our cells that processes nutrients into the energy cells need to survive.
“We have isolated the primary disease-causing component of muscular dystrophy to the mitochondrial permeability pore,” says the study’s corresponding author Jeffery Molkentin, PhD. “If we prevent this pore from functioning, dystrophic disease in the mouse models we studied almost completely vanishes. We see the protection lasting past one year of life in the mouse, which translates to about 40 years of life for a human.”
Molkentin is a widely respected expert in the basic science of muscle cell function and formation. He is co-executive director of the Heart Institute at Cincinnati Children’s and director of its Division of Cardiovascular Biology. He has studied muscular dystrophies for over 20 years.
Molkentin cautions that this discovery was achieved by observing outcomes in mice that were genetically modified to lack two genes that control mitochondrial permeability transition pore (MPTP) formation. Much more research beyond this early success will be needed to develop a safe and effective treatment for people with MDs.
Mitochondria take center stage
Mitochondria organelles are surrounded by their own membrane. However, when exposed to oxidative stress or a pathologic overload of calcium ions (Ca2+) mitochondria open a pore in their protective membrane. The influx of excess calcium causes the organelle to burst, which in turn causes muscle fibers to die, eventually leading to wasting of entire muscle groups.
This process of mitochondrial pore-regulated cell death has been observed in other conditions including heart muscle damage after heart attacks and neurodegenerative diseases including Huntington’s disease, Alzheimer’s disease, and amyotrophic lateral sclerosis (ALS).
In this study, Molkentin and colleagues reveal the mechanisms at work when this mitochondria-destroying process occurs in MDs. The team determined that two genes in mice—Slc25a4 and Ppif—work in concert to mediate unwanted pore formation. Controlling either one by themselves only slowed MD progression, but the absence of both together stopped it.
“We found direct evidence that these genes produce required components that govern cell death, which opens a previously unrecognized pathway for potentially treating MDs and other necrotic diseases,” Molkentin says.
Challenges ahead
Developing a medication to protect mitochondria in muscle cells will require much more study, Molkentin says.
The mouse gene Ppif produces a protein called CypD. Researchers established years ago that the immune-suppressing drug cyclosporin A can block this protein, but long-term use of the drug at high doses poses significant risk of side effects. In MD, the drug only mildly slows the disease in animal models.
Meanwhile, the mouse gene Slc25a4 encodes a protein called ANT1. There are no medications that target this protein. The existing compounds known to bind with this protein are fatally toxic, so new compounds would be needed.
“If a nontoxic ANT inhibitor can be identified, that also can target the mitochondrial pore, our results suggest that combined treatment with low dosages of a CypD inhibitor could be a novel therapeutic strategy,” Molkentin says. “Such a treatment could provide benefits independently or in combination with other gene therapies.”
Would such a treatment “cure” muscular dystrophy? It’s too soon to say. This study focused exclusively on skeletal muscle. More research would be needed to determine if the mitochondria-protecting approach also would protect against MD-related heart damage or other organ dysfunctions.
About this study
In addition to Molkentin, Cincinnati Children’s researchers involved in this study included first author Michael Bround
Funding sources included the National Heart, Lung, and Blood Institutes (R01HL132831 and R01HL150031), the National Institute of Arthritis and Musculoskeletal and Skin Diseases (K99AR078253), the L. B. Research and Education Foundation, and the Leducq Foundation.
| Original title: | ANT-dependent MPTP underlies necrotic myofiber death in muscular dystrophy |
| Published in: | Science Advances |
| Publish date: | Aug. 25, 2023 |
Research By

Our laboratory investigates a range of focus areas, all of which center on understanding the molecular mechanisms of heart and skeletal muscle disease.
Latest Posts


About this blog
The Research Horizons blog features news and insights about the latest discoveries and innovations developed by the scientists of Cincinnati Children's. This blog does not provide medical advice, diagnosis, or treatment. Email researchnews@cchmc.org with questions or ideas.
Subscribe to Our Newsletter