Inhibiting MEK Protein Shrinks NF1 Plexiform Tumors
Research By: Nancy Ratner, PhD | Brian Weiss, MD
Post Date: June 30, 2019 | Publish Date: Dec. 29, 2016

After five other medications fail to shrink NF1 tumors, selumetinib becomes first to demonstrate lasting improvement.
In some ways, the impressive results from a clinical trial published in The New England Journal of Medicine mark the triumph of a scientific career.
Decades of laboratory research leads to a breakthrough in understanding the cause behind a disfiguring, potentially life-threatening disorder. More work produces a genetically engineered mouse model that helps analyze potential treatments. Then finally, a phase 1 clinical trial reports benefits so clear that simple photographs can tell the story. Yet in other ways, this achievement marks just the beginning of the journey.
That’s the story behind the news that an MEK inhibitor called selumetinib demonstrates long-term benefits against the genetic disorder neurofibromatosis type 1 (NF1). It’s a success story built upon years of preclinical work led by Nancy Ratner, PhD, and Jianqiang Wu, MD, Division of Experimental Hematology and Cancer Biology, and carried forward by a small army of clinical colleagues across multiple medical centers.
NF1, A DISFIGURING CONDITION
NF1 affects approximately one in every 3,500 live births in the U.S. and affects more than 100,000 people in the U.S. Of those children, 20 to 50 percent develop significant plexiform neurofibroma (PN) tumors along their peripheral nerves.
PNs are not malignant, but they can be painful, disfiguring and dangerous. Depending on their location, they can cause blindness, disrupt growth, or damage other tissues. While surgeons can sometimes remove a part of these tumors, complete resection is rarely possible.
The NF1 Clinic at Cincinnati Children’s, headed by Elizabeth Schorry, MD, follows more than 1,200 children with the condition. Brian Weiss, MD, is an oncologist who treats many of these children. Both served as co-authors in the NEJM study.
“Until very recently, we had nothing beyond surgery that could shrink these tumors. Now, we finally have something effective,” Weiss says.
Until now, the trail toward an effective treatment had been littered with failure. Clinical trials had proven disappointing for five other drugs: sirolimus, tipifarnib, pirfenidone, imatinib and pegylated interferon alfa-2b. Selumetinib is the first to show significant benefit.
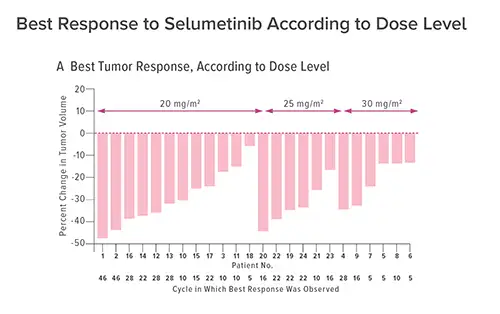
SELUMETINIB SHOWS RESULTS IN NF1 CLINICAL TRIAL
This clinical trial tracked 24 children who were enrolled between 2011 and 2014. The children were treated across a median number of 30 cycles (28 days each) with oral capsules of selumetinib taken every 12 hours.
In 17 children (71 percent) tumors shrank by at least 20 percent. Also, no progression occurred during a range of 23 to 46 cycles and a number of patients experienced reductions in pain, disfigurement and functional impairment.
Some of the improvements were dramatic. In one patient with a tumor so large that it caused severe hip disfigurement, 10 cycles of treatment had restored near-normal posture.
The NEJM paper included 23 co-authors, which reflects the sheer scale of this study. The National Cancer Institute (NCI) oversaw the clinical trial. Cincinnati Children’s, the NCI Pediatric Oncology Branch, Children’s Hospital of Philadelphia and Children’s National Health System recruited patients.
Senior author Brigitte Widemann, MD, of the NCI, secured an agreement from the pharmaceutical company AstraZenica to provide the medication. Other Cincinnati Children’s co-investigators on the study included Lindsey Aschbacher-Smith, MS, and Tilat Rizvi, PhD, who evaluated mouse tumors as part of the project.
LONG HOURS OF NEUROFIBROMATOSIS RESEARCH BEAR FRUIT
Ratner led the pre-clinical work that led to the trial, and Ratner and Wu developed the critical mouse model of neurofibroma tumors.
Ratner’s team used that mouse model to become the first to report (in 2013 in the Journal of Clinical Investigation) that blocking a single, critical protein (MEK) in the NF1 molecular process was effective at shrinking plexiform neurofibromas.
In addition to the clinical trial results, even more mouse data was presented in the NEJMstudy. In the mice, MEK inhibition occurred at two hours after drug administration. In addition, tumors shrank even when treatment was given for five days a week rather than seven.
“The clinical trial shows that a single agent given at low dose inhibits MEK for only a short time, yet significantly shrinks even very large plexiform neurofibromas,” Ratner says. “The study also demonstrates that our mouse model can accurately predict effects in humans, which means that we can use the model to test additional inhibitors that affect the MEK pathway.”
MORE TESTING PENDING FOR MEK INHIBITORS
At least four other MEK inhibitors are being tested in clinical trials, including work involving Cincinnati Children’s, Ratner and Weiss say. It is hoped that one of those drugs, by itself or combined with other agents, will produce even better results.
“Selumetinib may not be the answer, at least not by itself,” Weiss says. “It did shrink tumors for most participants, but it did not eliminate them. The drug also has to be taken long-term and that could mean it will eventually stop working.”
Ratner also found that MEK inhibitor effects are transient in her mouse model. She predicts that a combination therapy eventually will emerge.
“In my view, the next goal is to find a compound to add to an MEK inhibitor to see if we can melt these tumors completely.”
| Original title: | Activity of Selumetinib in Neurofibromatosis Type 1-Related Plexiform Neurofibromas |
| Published in: | New England Journal of Medicine |
| Publish date: | Dec. 29, 2016 |
Research By




Latest Posts
About this blog
The Research Horizons blog features news and insights about the latest discoveries and innovations developed by the scientists of Cincinnati Children's. This blog does not provide medical advice, diagnosis, or treatment. Email researchnews@cchmc.org with questions or ideas.
Subscribe to Our Newsletter