Ewing Sarcoma Tumors Can Be Split Into 2 Groups for Targeted Care
Post Date: November 17, 2022 | Publish Date: Nov. 17, 2022

Findings may improve management of the disease
Ewing sarcoma is a fast-growing form of bone and soft tissue cancer that strikes about 200 children and young adults per year in the United States.
Caught early, before the tumors have spread, the five-year survival rate for this cancer reaches 70-80%. However, over 25% of patients with initially localized disease and a much higher percentage of patients with initially metastatic disease have a relapse. Treatments have proven much less effective against metastatic or relapsed disease.
Now, a study led by experts at Cincinnati Children’s helps reveal why treatment outcomes can vary among patients and even in the same patient over the course of disease. Detailed findings were published Nov. 17, 2022, in Clinical Cancer Research, a journal of the American Association for Cancer Research.
The research team, led by corresponding author Rashmi Hegde, PhD, Division of Developmental Biology, reports finding two subgroups of Ewing sarcoma that respond differently to targeted drug therapy. The team also shows that both tumor groups can occur in the same person as disease progresses, suggesting that repeated biopsies may be needed over time to assure the most accurate treatment targeting.
Receptor location matters
Rapid cell growth in many types of cancer depends heavily on the insulin-like growth factor-1 receptor (IGF-1R) cell signaling pathway. But in Ewing sarcoma, tumors respond differently to drugs intended to block that pathway based on whether receptors are located in, or on, the cancer cell.
In one group of tumors, the Cincinnati Children’s team found these critical receptors concentrated within tumor cell nuclei, where IGF-1R promoted tolerance to DNA damage and made the tumor cells less vulnerable to treatment. But in another group, the receptors were located primarily in outer cell membranes, the tumor cells were under high levels of replication stress, and were more susceptible to treatment with the experimental compound Linsitinib.
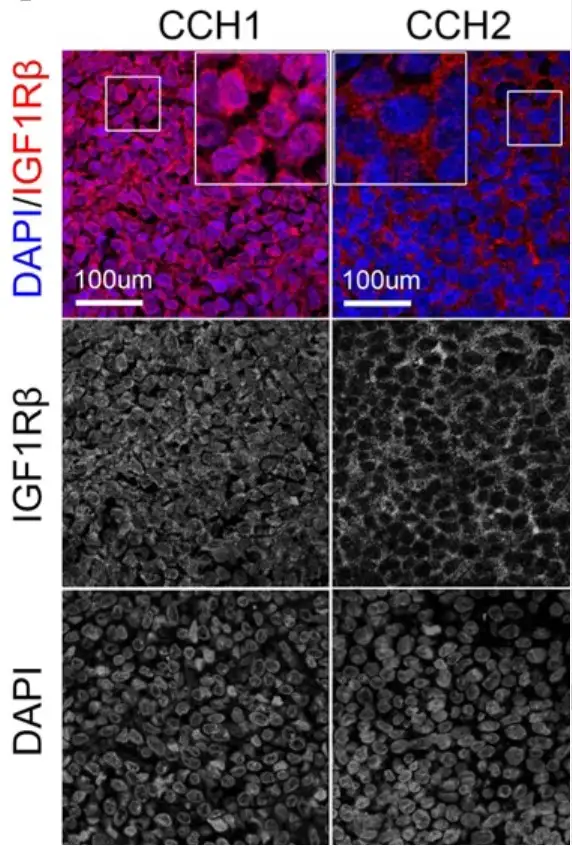
In mouse studies, and in lab tests involving human cells, the team did find a way to shrink tumors with nuclear IGF-1R. It required a combination of Linsitinib and another experimental drug called AZD1775. That second drug, part of a class of drugs known as checkpoint inhibitors, disrupts the function of the WEE1 protein, which helps regulate the timing of cell division.
“While the combined treatment approach shrank tumors from both groups of Ewing sarcoma tumors, the higher cost and potential for side-effects with combination therapies suggest that the two-drug approach may be of greater benefit to patients with nuclear IGF-1R containing tumors,” Hegde says.
Next step: refining biomarkers
Using current technology, care teams could conduct multiple biopsies over the course of disease management to determine the best course of treatment. But biopsies can be painful and come with risks. Less-invasive testing methods would be preferred.
“That means more research is needed to establish biomarkers that can be collected from blood or other less-invasive ways and still accurately distinguish between these classes of Ewing sarcoma tumors,” Hegde says.
About the study
In addition to Hegde, this study was led by three co-first authors: Upendra Soni, PhD, Yuhua Wang, PhD, and Ram Naresh Pandey, MS. Joseph Pressey, MD, Division of Oncology at Cincinnati Children’s, provided clinical insights and was a co-author. Collaborators also included experts from the Abigail Wexner Research Institute at Nationwide Children’s Hospital.
Funding sources for this work included the National Institutes of Health (RO1 CA207068) and a grant from the Jeff Gordon’s Children’s Foundation.
| Original title: | Molecularly defined subsets of Ewing Sarcoma tumors differ in their responses to IGF1R and WEE1 inhibition |
| Published in: | Clinical Cancer Research |
| Publish date: | Nov. 17, 2022 |


Latest Posts
About this blog
The Research Horizons blog features news and insights about the latest discoveries and innovations developed by the scientists of Cincinnati Children's. This blog does not provide medical advice, diagnosis, or treatment. Email researchnews@cchmc.org with questions or ideas.
Subscribe to Our Newsletter