Discovery Suggests Potential Non-Surgical Option for Children, Adults with Marfan Syndrome
Research By: Katherine Yutzey, PhD
Post Date: January 14, 2020 | Publish Date: Jan. 14, 2020
Thanks to decades of improving surgical techniques and medications to manage heart health, most people born with Marfan syndrome can expect to live a normal life span. However, nearly all face the likelihood of requiring open-heart surgery at some point.
Now a study led by experts at Cincinnati Children’s and published Jan. 14, 2020, in the journal Circulation, reports finding a new medication target that could reduce or delay the need for major surgery.
What is Marfan syndrome?
This mostly inherited genetic condition, discovered in the late 1800s, significantly affects how the body forms connective tissues, including in the aorta and heart valves. It occurs in one in every 5,000 to 10,000 births, which means doctors estimate that about 33,000 to 65,000 people in the US have this condition.
People with Marfan syndrome tend to grow tall and thin with unusually long limbs, fingers and toes. They experience a variety of skeletal and eye issues, but the most serious complications involve cardiovascular health. Many begin taking beta blockers and other heart medications early in childhood, and many eventually require surgical aortic root or mitral valve replacement or repair.
Related similar conditions include Loeys-Dietz syndrome and Ehlers-Danlos syndrome.
Tissue damaging process traced to resident immune cells
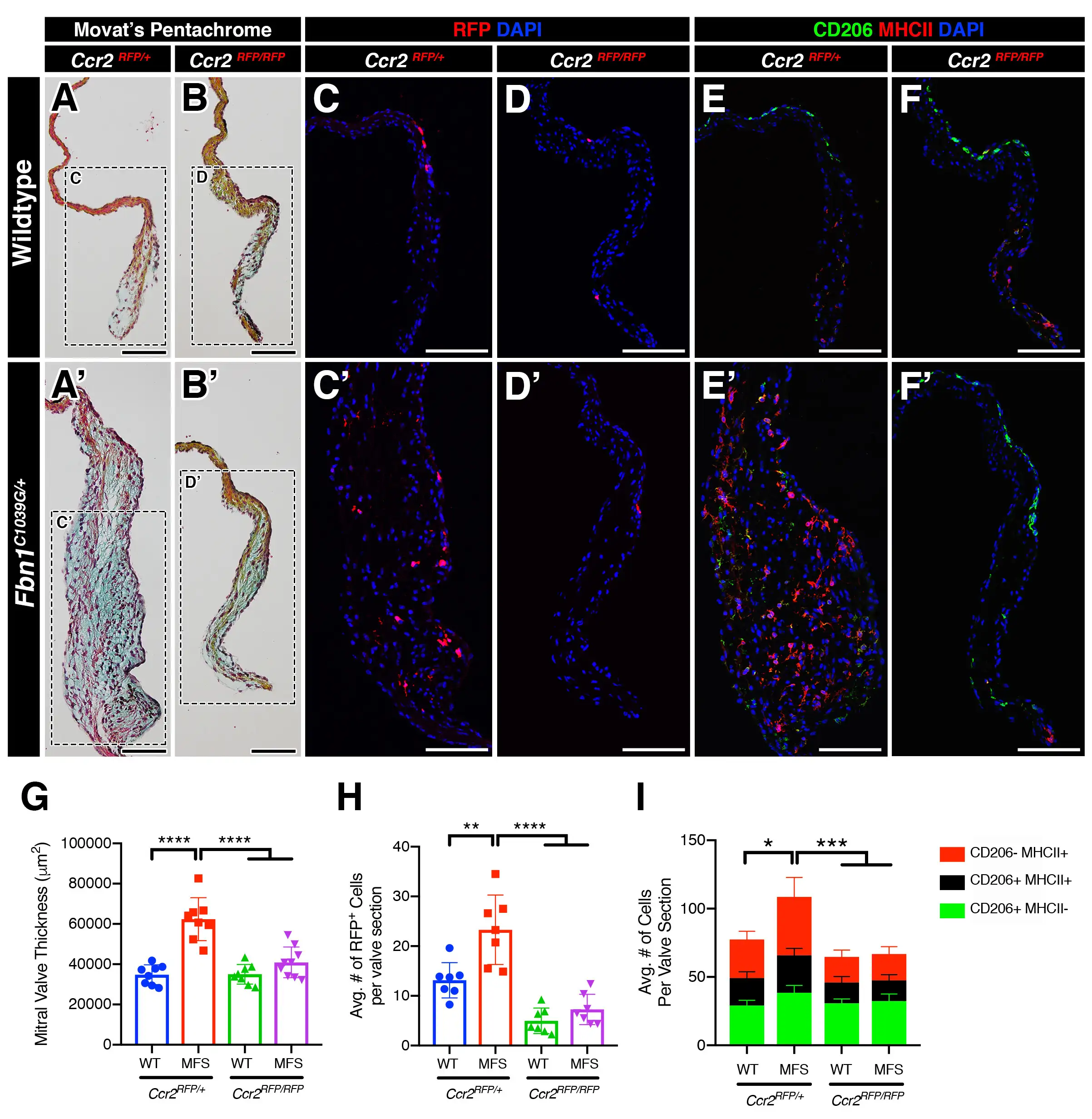
In the new study, Katherine Yutzey, PhD, of the Division of Molecular Cardiovascular Biology, led a team that explored how Marfan syndrome works at a cellular level. They were inspired by other recent research that had shown that mouse and human heart valves have immune cells built into their tissues.
The team determined that most of the increased numbers of immune cells in diseased valve tissue are C-C chemokine receptor type 2-positive (CCR2+) monocytes. These cells are recruited to the damaged tissue from the bloodstream to converge at the site of valve structural abnormalities.
But instead of performing their normal role of promoting healing, these monocytes wind up contributing to myxomatous valve degeneration (MVD), Yutzey says. Excessive monocyte infiltration generates excess inflammation, which in turn increases the progression of valve disease.
The researchers found this valve-degenerating process occurring in mice, larger mammals and in human heart tissue cultures.
“These valve defects in Marfan syndrome are the leading cause of death in infants with the syndrome and a major cause of morbidity and mortality later in life in these individuals,” Yutzey says.
Immune cell role suggests potential treatment
Upon learning that circulating monocytes were contributing to a distinct macrophage population in the murine heart valve, the team set out to determine whether blocking the monocyte activity would protect valve tissues from deterioration.
“Remarkably, inhibiting macrophage recruitment by CCR2 deficiency in monocytes was protective against MVD progression in MFS mice, resulting in valves showing minimal leaflet thickening and preserved matrix integrity,” the co-authors found. “Altogether, our results suggest sterile inflammation as a novel paradigm underpinning disease progression, and we identify, for the first time, monocytes as a viable candidate for targeted therapy in MVD.”
However, restricting CCR2+ macrophages did not offer protection against aortic aneurysms, another serious complication of Marfan syndrome. Diseased aortas in animals and human patients with Marfan syndrome did not have increased CCR2+ macrophages and, in mice, structural abnormalities of the aorta were not improved with loss of CCR2+ macrophages.
What’s next?
“There are drugs currently in clinical trials for inhibiting CCR2+ cells and we are currently testing them in our mouse model,” Yutzey says.
Those results, and other steps, will be needed before clinical trials can begin in humans, she says.
| Original title: | Deficiency of circulating monocytes ameliorates the progression of myxomatous valve degeneration in Marfan syndrome |
| Published in: | Circulation |
| Publish date: | Jan. 14, 2020 |
Research By

Latest Posts



About this blog
The Research Horizons blog features news and insights about the latest discoveries and innovations developed by the scientists of Cincinnati Children's. This blog does not provide medical advice, diagnosis, or treatment. Email researchnews@cchmc.org with questions or ideas.
Subscribe to Our Newsletter