3D Human Organoids Will Help Reveal Molecular Secrets of Deadly Liver Diseases
Research By: Takanori Takebe, MD | James Wells, PhD
Post Date: July 3, 2019 | Publish Date: May 30, 2019
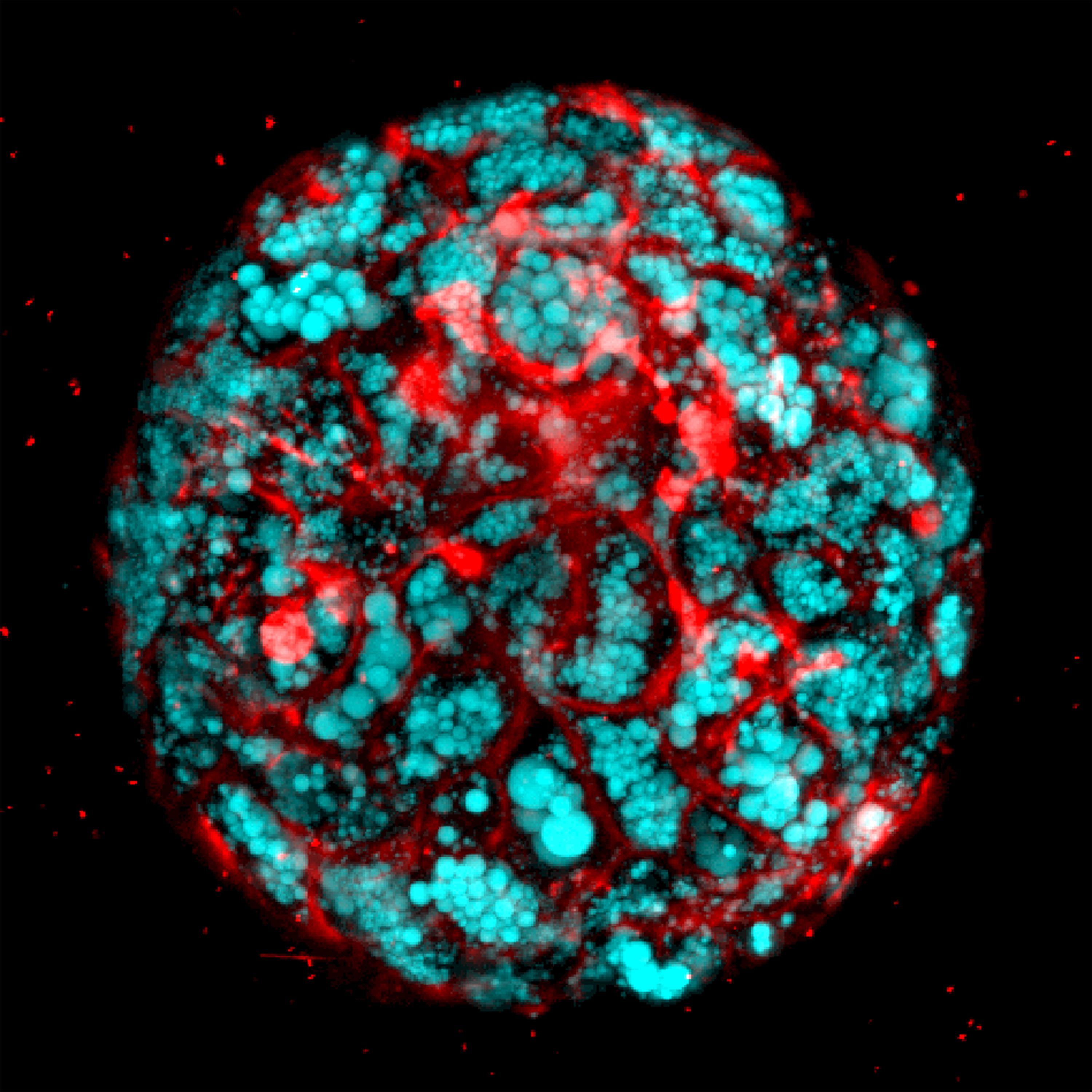
“These organoids generate different types of liver cells like hepatocytes, stellate and Kupffer-like cells, and they accurately recreate inflammation, fibrosis and other features of liver disease.’’
Scientists have successfully bioengineered human liver organoids that faithfully mimic key features of fatal liver disease in the laboratory. This allowed them to uncover underlying disease biology in the organoids and test a potential therapy that in preclinical lab tests reversed an often-fatal childhood condition called Wolman disease.
In findings published online by the journal Cell Metabolism, researchers at Cincinnati Children’s Hospital Medical Center say their study overcomes major hurdles to unraveling the molecular mysteries of liver diseases and finding desperately needed new therapies. It also leads to personalized methods to study inflammation and fibrosis in liver disease that match the unique genetics and biology of individual patients, according to study authors.
“Although current human organoid systems can recreate organ architecture in living lab organisms like mice, they fail to capture the complex pathologies of inflammation and fibrosis in liver diseases,” said Takanori Takebe, MD, lead study investigator and a physician at the Cincinnati Children’s Center for Stem Cell and Organoid Medicine (CuSTOM).
“We developed a reproducible method to bioengineer complex, multicellular human liver organoids using pluripotent stem cells derived from healthy human donors and those with liver disease,” he said. “These organoids generate different types of liver cells like hepatocytes, stellate and Kupffer-like cells, and they accurately recreate in the lab inflammation, fibrosis and other features of liver disease.’’
That is significant because years of attempted drug development for conditions like fatty liver disease (NASH, NAFLD, etc.) and Wolman have been hindered by a lack of human disease models that accurately mimic the diseases, Takebe said. And although certain animal models like mice are available, they aren’t always reliable for studying human disease.
The study is a collaboration of scientists at Cincinnati Children’s, the Institute of Research at Tokyo Medical and Dental University (TMDU) in Japan (where Takebe is also a member of the medical staff), and researchers at four other institutions in Japan and the United States.
Often Silent Killers
One challenge of battling liver disease is it can be a silent killer, producing few initial symptoms that might prompt early, proactive medical treatment.
Wolman disease is a genetic disorder that results in deficiency of an enzyme called lysosomal acid lipase (LAL), which breaks down fats in the body, according to the National Institutes of Health. Without the enzyme, fats over-accumulate in liver tissues and cause bloating, swelling of the stomach, vomiting and an enlarged liver or spleen, leading to life-threatening complications.
In nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH), fat accumulation, inflammation and fibrosis cause serious harm to the liver, an organ that helps the body digest food and filter out toxic substances. The causes of NAFLD and NASH remain under investigation, although research points to certain health conditions that make fatty liver disease more likely, such as obesity or insulin resistance. Research also points to exposure to certain medications, toxins or diseases as potential but less common causes.
Finding Possible Solutions
In this study, Takebe and colleagues used 11 different lines of healthy and diseased pluripotent stem cells to generate complex multi-cellular human liver organoids. The diseased pluripotent cells included those grown from donors with a deficiency in the LAL protein.
The researchers treated the organoids with free fatty acids—byproducts of fat metabolism in fatty tissue. This helped drive liver fat accumulation, inflammation and fibrosis. The organoids were able to recreate liver disease in the lab in a step-by-step, successive manner similar to Wolman disease in children.
As liver disease developed in the organoids, researchers used an extremely high-resolution imaging technology called atomic force microscopy. This allowed them to demonstrate and observe progressive levels of organ stiffening that they said confirms the increasing severity of accumulating liver fibrosis.
After concluding that Wolman disease in the human organoids was caused by LAL deficiency, the researchers were able to reverse that deficiency by exposing the organoids to a recombinant LAL protein. Next, study authors leveraged earlier research that shows LAL deficiency causes over activation of a protein called mammalian target of rapamycin (mTOR), which is linked to liver fibrosis.
Previous research also shows that mTOR can be suppressed by a receptor protein called farnesoid X receptor (FXR), which functions in the nucleus of intestinal and liver cells. FXR upregulates and down-regulates genes that control fat metabolism and bile acid processes in the liver. Takebe and his colleagues decided to treat the human liver organoids with Wolman disease with an FXR agonist called obeticholic acid. FXR is an acid that synthesizes a protein called fibroblast growth factor 19 (FGF19) in the ileum of the small intestine.
FXR treatment suppressed fat accumulation in the human liver organoids and prolonged their survival. This opens the possibility of further preclinical testing to see whether or not FXR treatment might be an appropriate strategy for treating Wolman disease, according to study investigators. Because the study’s findings are preclinical and involve laboratory models, additional research is needed to show whether the findings will apply to clinical treatment in patients.
—By Nick Miller
| Original title: | Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids |
| Published in: | Cell Metabolism |
| Publish date: | May 30, 2019 |
Research By


Latest Posts



About this blog
The Research Horizons blog features news and insights about the latest discoveries and innovations developed by the scientists of Cincinnati Children's. This blog does not provide medical advice, diagnosis, or treatment.
Subscribe to Our Newsletter